

Glyco-bridge strategy enhances CAR-T cell killing of Tn-MUC1-expressing cancer cells
Tn antigens are overexpressed due to the upregulation of ppGalNAc-Ts, genetic silencing or mutation of C1GALT1, or mutations in COSMC43,44,45,46,47,48,49,50 (Fig. 1a). Tn antigens decorate the backbone of cell-surface glycolipids and glycoproteins, such as cell-surface mucins. Notably, cancer-associated mucins are overexpressed in tumors and cell lines and are associated with high heterogeneity at the cell surface level54,55. Since the mucins can form a nanoscale glycocalyx barrier at the cellular interface that impedes immune cell interaction, we tested the hypothesis that enhancing CAR T cell binding to cancer cells could help overcome this barrier. Building on previous approaches that utilized two CAR constructs56,57,58, we designed our CAR construct with a non-signaling bridge system. In contrast to prior methods, we employed a distinct transmembrane domain to prevent heterodimerization and removed the intracellular domain from the bridge construct. We used this modification with the hypothesis that the bridge would overcome the glycocalyx barrier solely by facilitating binding of the CAR T cell to the cancer cell (Fig. 1b). Our glyco-bridge was designed to engage the cancer-associated, cell surface mucin structure Tn-MUC1 with a single-chain variable fragment (scFv). We initially tested a Tn-MUC1 scFv (5E5)23 as the glyco-bridge binder alongside a mesothelin-targeting CAR molecule (with an SS1 scFv)59 and used a CD19 scFv (FMC63)60 bridge as a control, since CD19 is not expressed by solid tumor cells. All of these scFvs have been used in CAR T cells that have either entered clinical trials or have received FDA approval2,23 and contain a (G4S)3 linker between their variable heavy and light chains. For the CAR construct, the SS1 scFv was directly fused to a CD8 hinge/transmembrane domain, a 4-1BB costimulatory domain, and CD3ζ (Fig. 1c). The bridge constructs incorporated a CD28 hinge/transmembrane domain to prevent dimerization with the CAR molecule. To better anchor the bridge to the cell membrane, we incorporated CD28 intracellular domain that we mutated in its key subdomains to prevent signaling. Specifically, we mutated proline residues P208 and P211 to alanine within the PRRP subdomain to prevent SH3-containing proteins (such as Itk and Tec) from binding, and we modified the PYAP motif by substituting tyrosine (Y191) with phenylalanine, inhibiting binding to the SH2 domain of Lck, a key mediator in T-cell signaling61 (Fig. 1b). Expression of the CAR and glyco-bridge molecules was confirmed using an ALFA tag and the (G4S)3 linker (Fig. 1d). In both constructs, the transduction rate of CAR molecules ranged from 40 to 50% and remained stable over time. The CD19-bridge alongside the CAR molecule demonstrated strong binding affinity to recombinant CD19 protein (Supplementary Fig. 1a).

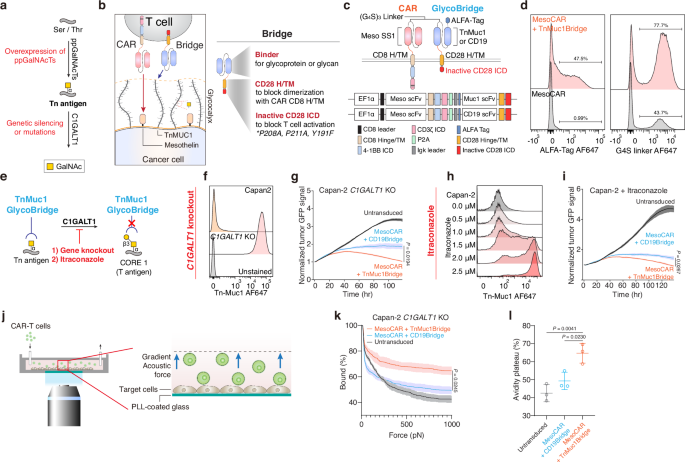
a Biosynthetic pathway for O-glycans with polypeptide N-acetylgalactosaminyltransferases (ppGalNAcTs) and C1GALT1 gene. b Schematic of mesothelin-targeting Chimeric Antigen Receptor (CAR)-T cells with glyco-bridge. c Graphic representation of the constructs used to make CAR-T cells with Tn-MUC1 bridge or CD19-bridge. The glyco-bridge contains a CD28 hinge and transmembrane domain (H/TM) and an inactive CD28 intracellular signaling domain (ICD). d Flow cytometry analysis of glyco-bridge surface levels on T cells using ALFA-Tag (left) and (G4S)3 linkers expression levels (right). e Schematic of the biosynthetic pathway for Tn antigen. f Flow cytometry analysis of Tn-MUC1 surface level on Capan-2 and Capan-2 C1GALT1 KO cells. g Representative real-time cytotoxicity assay against Capan-2 C1GALT1 KO cells at a 1:1 E:T ratio (relative to day 0 tumor seeding) from n = 3 distinct human blood cell donors. Results are mean ± s.d. of n = 3 independent measurements. h Surface level of Tn-MUC1 on Capan-2 following itraconazole treatment at the indicated concentrations for 48 h. i Representative real-time cytotoxicity assay against 2.5 µM itraconazole-treated Capan-2 cells at a 1:1 E:T ratio (relative to day 0 tumor seeding) from n = 3 three distinct human blood cell donors. Results are mean ± s.d. of n = 3 independent measurements. j Schematic of cell avidity measurement with acoustic force microscopy. k Strength of interaction between Capan2 C1GALT1 KO target cells and CAR-T cells expressing TnMUC1 or CD19-bridges. Percentage of total CAR-T cells remaining bound to target cells as the acoustic force ramp is applied from 0 to 1000 pN are shown. Results are mean ± s.e.m. of n = 3 independent measurements. l Percentage of CAR-T cells remaining bound to the target cells at the avidity plateau under 1000 pN force from Fig. 1k. Results are mean ± s.d. of n = 3 independent measurements. In d, f, h, representative flow cytometry data from three independent experiments. In g, i, and k, statistical analysis was performed by two-way ANOVA with correction for multiple comparisons. In l, statistical analysis was performed by one-way ANOVA with Tukey’s post hoc tests.
We used the Capan-2 pancreatic ductal adenocarcinoma (PDAC) cell line, which expresses high levels of cell-surface mucins, including MUC1 (Supplementary Fig. 1b), as target tumor cells. This cell line has previously been used in studies evaluating 5E5 CAR-T cells targeting Tn-MUC1, demonstrating functional recognition and cytotoxicity23. They also express high levels of mesothelin (the target of the CAR) and lack CD19 expression (the target of the control bridge). To further increase the surface expression of Tn-MUC1 for proof-of-concept evaluation of the glyco-bridge strategy, we generated C1GALT1 knockout Capan-2 cells using CRISPR/Cas9-mediated gene editing or treated the cells with itraconazole, an antifungal drug, to inhibit C1GALT1 enzymatic activity62 (Fig. 1e). Both Capan-2 C1GALT1 KO cells and itraconazole-treated Capan-2 cells had increased Tn-MUC1 expression. In a real-time killing assay with Capan-2 C1GALT1 KO cells, mesothelin-targeted CAR-T cells incorporating a Tn-MUC1-bridge demonstrated higher killing efficiency compared to those with the control CD19-bridge (Fig. 1f, g and Supplementary Fig. 1c). Likewise, combining itraconazole treatment with Tn-MUC1 bridge CAR-T cells further enhanced killing of Capan-2 cells in vitro (Fig. 1h, i). To determine if this effect would be applicable to either bridge molecule, we overexpressed CD19 in Capan-2 cells. The CD19-bridge enhanced the cytotoxicity of mesothelin-targeted CAR-T cells. To test this concept further, we utilized the RPMI 8226 multiple myeloma cell line, which also exhibits high expression of MUC1 but lacks detectable mesothelin and CD19. After CD19 overexpression, we observed a similar enhancement in BCMA-targeted CAR-T cell cytotoxicity, suggesting that the bridge strategy is compatible with different CAR constructs and tumor types. (Supplementary Fig. 1d–g).
To determine whether bridge expression changed the phenotype of CAR-T cells, we analyzed CAR-T cells on day 14 after CD3/CD28 bead-based T cell activation and classified them as naïve (CD45RA+ CCR7+), terminally differentiated effector (TEMRA; CD45RA+ CCR7−), central memory (TCM; CD45RA− CCR7+), or effector memory T cells (TEM; CD45RA− CCR7−) within the CD4 and CD8 CAR-T cell populations. There were no differences in cell phenotypes between the glyco- and CD19-bridge CAR T cells (Supplementary Fig. 1h, i). Furthermore, there was no significant difference in the overall cytokine secretion between the two bridge conditions after 24-h stimulation with Capan-2 C1GALT1 KO cells. However, TNF-α levels were modestly higher with the TnMUC1 bridge, although this difference did not reach statistical significance. (Supplementary Fig. 1j–m).
To test the hypothesis that the bridge molecule enhanced T cell adhesion to the target cells, we measured the cell avidity of the Tn-MUC1 bridge against the Capan-2 C1GALT1 KO cell line. Cells were seeded in z-Movi Cell Avidity Microfluidics chips to form a monolayer on the surface. CAR-T cells with bridges were then introduced and allowed to bind to the target cells. CAR-T cells were tracked, and the binding avidity between target cells and CAR-T cells was quantified, with gradient acoustic forces applied to disrupt the interaction between the cells (Fig. 1j). We found that the Tn-MUC1 bridge significantly increased cell avidity compared to the control CD19-bridge against Capan-2 C1GALT1 KO cells. This suggests that the enhanced cytotoxic efficacy of the glyco-bridge CAR T cells in vitro is attributed to the increased avidity between the CAR T cell and target cell (Fig. 1k, l).
Tn-MUC1 bridge CAR-T cells enhance tumor suppression in Capan-2 mouse models
To prepare to test this approach in vivo, we first examined the impact of C1GALT1 KO and itraconazole treatment on Capan-2 tumor growth. Tumor cells (5 × 106) were subcutaneously injected into NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice, and itraconazole was administered via intraperitoneal injection at a dose of 40 mg/kg daily for 60 days. This dosage was carefully determined to balance therapeutic efficacy and potential toxicity concerns during long-term treatment, corresponding to clinically relevant plasma concentrations (0.5–1.0 μg/mL) observed in human patients receiving standard oral itraconazole therapy63,64,65. Overall, no significant differences in tumor growth were observed among the conditions. (Fig. 2a and Supplementary Fig. 2a, b). Consistent with in vitro findings, in vivo itraconazole treatment increased Tn antigen expression in tumors, with a 2.4-fold increase compared to Capan-2 cells. Similarly, C1GALT1 KO Capan-2 exhibited a 4.1-fold increase in Tn antigen expression compared to Capan-2 cells (Fig. 2b, c). We also confirmed that itraconazole had no effect on tumor growth at concentrations up to 10 μM in vitro, although Capan-2 C1GALT1 KO cells exhibited slower in vitro growth rates compared to wild-type cells (Supplementary Fig. 2c).

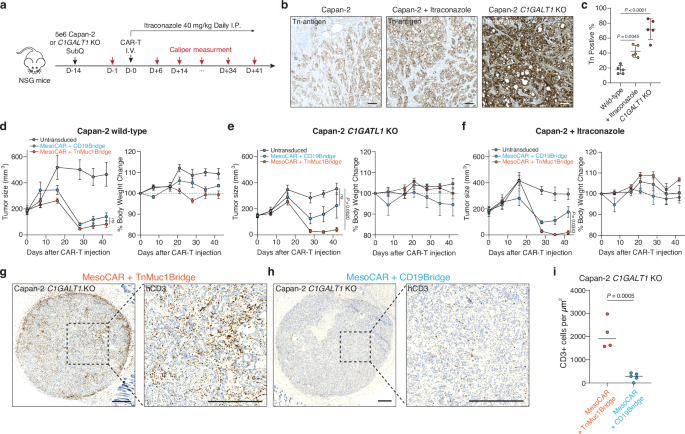
a NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice were subcutaneously (SubQ) injected with 5 x 106 Capan-2 or Capan-2 C1GALT1 KO cells on day −14. The mice were further treated intraperitoneally (I.P.) with itraconazole (40 mg/kg) once daily for 30 days. On Day 0, 5 mice per group received 2 × 106 CAR T cells for the Capan-2 C1GALT1 KO group or 2.5 × 106 CAR T cells for the Capan-2 group. CAR-T cells are the indicated mesothelin-targeted CAR with glyco-bridge or untransduced T cell. b Representative IHC images for Tn antigen in formalin-fixed, paraffin-embedded specimens from mice injected with Capan-2, Capan-2 C1GALT1 KO, and Capan-2 with itraconazole treatment. Scale bar, 10 μm. c Quantification of Tn antigen+ cells in tumors from each mouse. Results are mean ± s.d. of n = 5 mice. Tumor growth curve (left) and body weight change (right) for Capan-2 (d), Capan-2 C1GALT1 KO (e), and Capan-2 with itraconazole treatment (f). Results are mean ± s.e.m. of n = 5 mice. Representative IHC images for CD3 (T cell) in formalin-fixed, paraffin-embedded tumor specimens from mice injected with Capan-2 C1GALT1 KO and mesothelin-targeted CAR-T cells with Tn-MUC1 bridge (g) or CD19-bridge (h). Scale bar, 500 μm. i Quantification of infiltrating CD3+ cells in tumors of mice treated with mesothelin-targeted CAR-T cell using either a Tn-MUC1 or CD19 bridging strategy. Data represent the mean for n = 4 for mesothelin-targeted CAR-T cells with Tn-MUC1 bridge and n = 5 for CD19-bridge, with data from tumor-cleared mice excluded. Statistical analysis was performed by two-tailed t-tests. In c, statistical analysis was performed by one-way ANOVA with Tukey’s post hoc tests. In d–f, statistical analysis was performed by two-way ANOVA with correction for multiple comparisons.
Next, we measured CAR-T cell-mediated killing against Capan-2, Capan-2 C1GALT1 KO, and itraconazole-treated Capan-2 in vivo. Capan-2 cell lines (5 x 106) were injected subcutaneously into NSG mice. The tumors were monitored for 14 days until the average tumor size reached approximately 150–200 mm3. To determine the optimal dose, we tested mesothelin-targeted CAR-T cells at doses of 1 x 106 cells, 4 x 106 cells, and two separate infusions of 4 x 106 cells per week against Capan-2 C1GALT1 KO cells. Based on these experiments, we identified the optimal dose range (1–4 x 106 cells) for the Capan-2 model (Supplementary Fig. 2d). As expected, due to the relatively low levels of MUC1 expression at baseline, no significant difference in tumor size was observed between the two bridges in the CAR-T cell condition in the Capan-2 wild-type model (Fig. 2d). However, in the Capan-2 C1GALT1 KO model or the Capan-2 model treated with itraconazole, where MUC1 levels were increased, CAR-T cells with Tn-MUC1 bridge exhibited significantly more anti-tumor activity compared to the CD19-bridge (Fig. 2e, f and Supplementary Fig. 2e). Furthermore, no significant weight loss or poor body condition was observed (Fig. 2e, f). To further evaluate the effect of the glyco-bridge on CAR-T cell infiltration throughout the tumor, we stained human CD3+ cells in the residual tumors on Day 52 using an anti-human CD3 antibody. Notably, CAR-T cells with the Tn-MUC1 bridge showed approximately 8-times higher infiltration than the CD19-bridge into Capan-2 C1GALT1 KO tumors (Fig. 2g–i). The tumor control by mesothelin-targeted CAR-T cells with the glyco-bridge strongly correlated with tumor infiltration by the T cells. Taken together, these results support the use of a glyco-bridge with CAR molecules to enhance anti-tumor activity.
Activation of the CAR is dependent on recognition of the bridge’s antigen
Since we observed no significant difference in efficacy between the glyco- and CD19-bridge in tumors with relatively low Tn-MUC1 expression or CD19 expression, we next investigated how the expression level of the bridge target antigen contributes to CAR-T cell-mediated killing. As a working hypothesis, we tested four conceptual models of CAR-T cell-mediated killing based on the density of the antigen recognized by the bridge molecule, rather than the CAR itself: independent, insensitive, linear, and ultrasensitive. These models reflect different hypothetical dependencies of CAR-T cell activity on bridge antigen density. (Fig. 3a). As a model, we utilized the RPMI 8226 multiple myeloma cell line, which lacks expression of mesothelin and CD19 (Supplementary Fig. 3a, b) and has moderate expression levels of MUC1 (Fig. 3b). This made it an ideal system to test the contribution of the glyco-bridge, since CAR-T cells targeting mesothelin would not engage the tumor cells directly. Similar to Capan-2 cells, RPMI 8226 cells exhibited increased surface expression of Tn-MUC1 and Tn antigens upon itraconazole treatment, enabling us to assess how modulation of bridge antigen density impacts CAR-T cell activity. (Fig. 3b and Supplementary Fig. 3c). Using Tn-MUC1 bridge mesothelin-targeted CAR T cells against the RPMI 8226 cell line, there was no significant cytotoxicity, even with itraconazole treatment, suggesting that the Tn-MUC1 bridge was not involved in CAR-mediated killing when the expression of the CAR antigen was low (Fig. 3c). This suggested that sufficient activation of the CAR is necessary for the bridge to contribute to effective cytotoxicity. To verify this result with our other bridge molecule targeting CD19, we again used the RPMI8226 cells but overexpressed CD19 and compared them to wild-type cells with low CD19 expression. This time, the mesothelin-targeted CAR-T cells with the CD19-bridge exhibited increased cytotoxicity when CD19 was overexpressed (Fig. 3d–g). The same trend was observed in Jeko-1 cells, which similarly express low mesothelin but high levels of CD19 in WT cells (Supplementary Fig. 3a, b). Conversely, we knocked out CD19 from Jeko-1 cells by CRISPR/Cas9, then re-expressed CD19 by transduction generated clones with varying CD19 expression levels by single-cell sorting. The number of CD19 molecules per cell was quantified using the BD Quantibrite™ Kit (Fig. 3h). In Jeko-1 cells, CD19-bridge-mediated cytotoxicity was abrogated when CD19 was knocked out, demonstrating that the cytotoxicity was dependent on expression of the antigen targeted by the bridge molecule (Fig. 3i, j). Our data revealed a clear power-law dependence of CD19-bridge-mediated cytotoxicity on CD19 density in Jeko-1 cells, with a scaling exponent of −0.74 (Fig. 3k). Similarly, CD19 overexpression in RPMI-8226 multiple myeloma cell lines exhibited comparable trends, with a scaling exponent of −0.16 (Fig. 3g). Thus, these findings suggest that additional binding via the bridge molecule may contribute to CAR-T cell cytotoxicity even in the presence of minimal CAR antigen expression, raising the possibility of partial CAR target-independent killing.

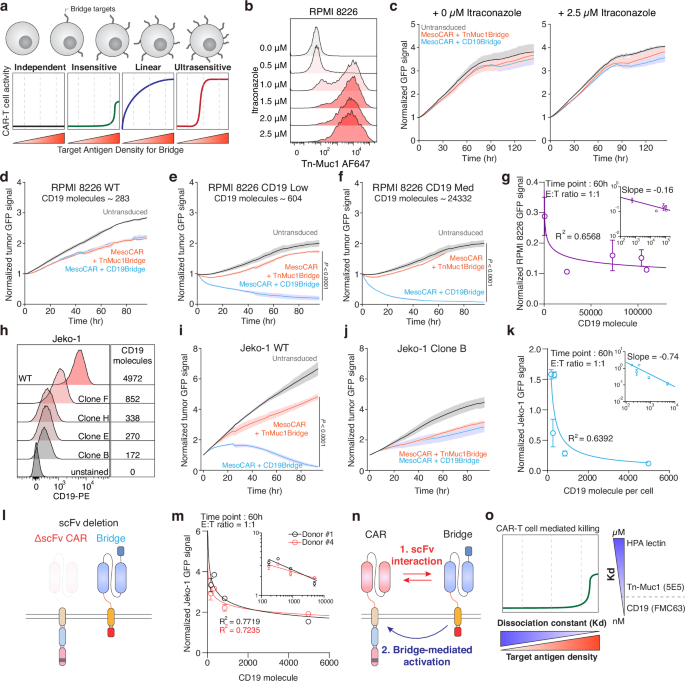
a Schematic showing the mechanism of glyco-bridge depending on the target antigen density of the glyco-bridge. b Surface level of Tn-MUC1 on RPMI 8226 cells following itraconazole incubation for 48 h. c Real-time cytotoxicity assay against RPMI 8226 cells with itraconazole with indicated bridges at a 1:1 E:T ratio. Results are mean ± s.d. of n = 3 independent measurements. d–f, Real-time cytotoxicity assay with mesothelin-targeted CAR-T cells with indicated bridges at a 1:1 E:T ratio. Panels show RPMI 8226 (d), low CD19 (e), and medium CD19 (f). Results are mean ± s.d. of n = 3 independent measurements. g log of the number of CD19 molecules versus log of the normalized target cell GFP signal from d–f at time point 60 h; best-fit line to a power-law model with slope = −0.16 and R2 = 0.6568. h Surface level of CD19 on Jeko-1 clones. i, j Real-time cytotoxicity assay with mesothelin-targeted CAR-T cells with indicated bridges at a 1:1 E:T ratio. Panels show Jeko-1 (i) and Jeko-1 clone B (j). Results are mean ± s.d. of n = 3 independent measurements. k log of the number of CD19 molecules versus log of the normalized target cell GFP signal from (i, j) at time point 60 h; best-fit line to a power-law model with slope = −0.74 and R2 = 0.6392. l Schematic showing ΔSS1 scFv on CAR molecules. m log of the number of CD19 molecules versus log of the normalized GFP signals from Jeko-1 cell lines from (h) using ΔSS1 scFv CAR at time point 60 h; best-fit line to a power-law model for each donor; R2 for 0.7719 (donor #1) and 0.7235 (donor #4). Results are mean ± s.d. of n = 3 independent experiments per donor. n Proposed relationship between CAR and bridge molecules. o Proposed the mechanism between glyco-bridge-mediated killing, target antigen density for the glyco-bridge, and binding affinity. In b, h representative flow cytometry data from three independent experiments. In e, f, and i statistical analysis was performed by two-way ANOVA with correction for multiple comparisons.
To further explore this CAR target-independent killing, we altered the different functional domains of the CAR and the bridge to determine which domains were necessary for T cell killing. We generated an scFv-deleted CAR, a complete CAR deletion, and a CD3ζ deleted-CAR. We also substituted the CD28 H/TM domain with the PDGFRβ TM domain to investigate the linkers interaction and the interaction between the endogenous TCR and CD28 or CD8. Additionally, we replaced the inactive CD28 domain with the NGFR ICD domain, which is unrelated to CAR signaling (Fig. 3l and Supplementary Fig. 3d). We used each of these CAR T cells against WT and CD19-overexpressing RPMI 8226 cells. Similar to what we had observed previously, there was no difference in the killing of WT RPMI 8226 cells. With CD19-overexpressing RPMI 8226 targets, only CAR-T cells containing intracellular signaling domains showed enhanced killing, confirming that bridge-mediated killing is CAR-dependent. Importantly, CAR constructs lacking the scFv still showed cytotoxicity in the presence of the CD19 bridge, suggesting that bridge can activate CAR signaling independently of direct antigen recognition by the CAR scFv (Fig. 3m and Supplementary Fig. 3e–h). Fluorescent microscopy also revealed no direct killing by the bridge alone, but it did show aggregation of Jeko-1 cells in the presence of the CD19-bridge. This clustering is consistent with bridge-mediated cell adhesion, likely resulting from bridge molecules binding to CD19 on neighboring target cells (Supplementary Fig. 3i). Based on these results, we identified two main interactions between the CAR molecule and the bridge molecule (Fig. 3n). Previous studies have shown that scFv aggregation occurs through VH/VL interactions alone, without protein unfolding, and this phenomenon is currently considered in CAR manufacturing to minimize scFv interactions66,67. Likewise, the bridge molecule can activate the CAR molecule to kill target cells.
We showed that CAR-mediated killing does not occur via the Tn-MUC1 bridge in Fig. 3c, whereas CAR-mediated killing can occur in a target-independent manner through the CD19-bridge. This finding led us to examine the affinity of the two scFvs. The dissociation constant of the FMC63 scFv (αCD19) ranges from 300 pM to 5 nM68,69,70, whereas the 5E5 scFv (αTn-MUC1) ranges from 2 to 4 nM71,72. This difference suggests that the ligand affinity, in addition to the antigen density, could play a role in the efficacy of killing mediated by the bridge. We hypothesize that despite their ability to engage target glycans, low-affinity binders like lectins may not provide sufficient binding strength to trigger CAR-mediated killing through the bridging interaction (Fig. 3o). Therefore, this approach would avoid undesirable CAR T cell activation in response to the bridge target alone, since lectins are also expressed in normal tissue (albeit at lower levels).
HPA lectin-based CAR-T cells expand the targeting scope of Tn antigens
To universally target Tn antigen structures beyond Tn-MUC1, we leveraged the HPA lectin as the antigen-binding domain of the CAR molecule. Unlike MUC1-specific binders, HPA recognizes the Tn antigen motif (GalNAcα1-Ser/Thr) across a wide range of glycoproteins, including mucins such as MUC5AC and MUC16 in pancreatic cancer73. This enables broader detection of Tn antigen-expressing tumor cells regardless of the specific mucin backbone. This lectin has a relatively low dissociation constant while maintaining high specificity for Tn antigen. The dissociation constant of HPA with GalNAc is approximately 130 µM74, which is lower than that of conventional FMC63 scFv. We firstly aimed to quantitatively evaluate the effectiveness of HPA as a binder for CARs. As previously reported44,75, the Tn structure is predominantly expressed in the ductal regions of pancreatic ductal adenocarcinoma76,77. We confirmed that HPA binds to these regions using HPA-biotin on a human pancreatic tissue microarray (PA807). H-score analysis showed significantly elevated HPA-biotin binding in PDAC tissues relative to normal pancreas, indicating increased expression of Tn antigen in PDAC patients. (Fig. 4a–c). Furthermore, when we inhibited C1GALT1 enzymatic function using itraconazole, HPA binding to Capan-2 cells increased compared to baseline (Fig. 4d). We then engineered a HPA-based CAR by replacing the scFv binder with HPA to determine its efficacy as a binding component (Fig. 4e). Since wild-type HPA has a hexagonal structure formed by two trimeric subunits, we designed a dual HPA (HPAx2) and triple (HPAx3) structure to enhance binding affinity. Each HPA unit is fused via a (G4S)3 linker (Fig. 4e).

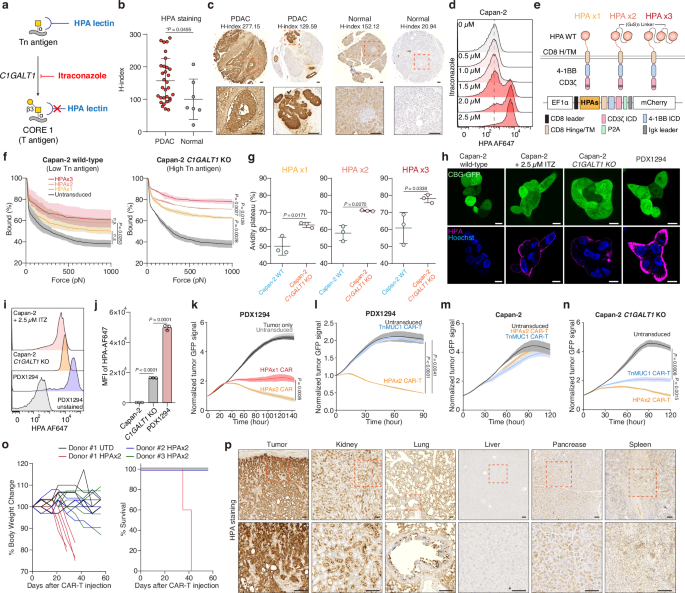
a Biosynthetic pathway for Tn antigen. b HPA staining quantified by H-score in PDAC versus normal pancreatic tissues (PA807; n = 31 and 7). H-score represents the weighted sum of cells with low (0.14–0.4), moderate (0.4–0.6), or strong (>0.6 OD) intensity. Significance was assessed by unpaired two-tailed t-test. c Representative IHC images of HPA-stained tissue sections from b. Scale bar, 100 μm. d Surface level of HPA binding on Capan-2 following itraconazole incubation at the indicated concentrations for 48 h. e Schematic showing HPA-based CAR T cells. f Strength of interaction between target cells and indicated HPA CAR-T cells. Percentage of total CAR-T cells remaining bound to target cells as the acoustic force ramp is applied from 0 to 1000 pN are shown. Results are mean ± s.e.m. of n = 3 independent measurements. g Percentage of CAR-T cells remaining bound to the target cells at the avidity plateau under 1000 pN force from Fig. 4f. Significance was assessed by unpaired two-tailed t-test. h Representative confocal images of HPA-stained cells. Scale bar, 10 μm. i Surface level of HPA binding on indicated target cells. j HPA binding on each cell from (g). Results are mean ± s.d. of n = 3 independent measurements. Real-time cytotoxicity assay against PDX1294 (k, l), Capan-2 (m), Capan2- C1GALT1 KO (n) target cells with indicated CAR T cells at 1:1-2:1 E:T ratio. Results are mean ± s.d. of n = 3 independent measurements. o NSG mice implanted s.c. with 5 × 106 Capan-2 C1GALT1 KO cells received 2 × 106 dual-HPA CAR-T or UTD cells on day 0 (5 mice/group; n = 3 donors). Body weight change (left) and survival percentage for n = 5 mice. p Representative HPA staining of tumor and major organs from o (n = 5 mice). Scale bar, 100 μm. In (d, i) representative flow cytometry data from three independent experiments. In f and k–n statistical analysis was performed by two-way ANOVA with correction for multiple comparisons. In (j), one-way ANOVA with Tukey’s post hoc tests.
To quantify the binding affinity of each HPA-based CAR, we measured cell avidity using the z-Movi Cell Avidity Microfluidics System against Capan-2 wild-type cells (low Tn antigen) and Capan-2 C1GALT1 knockout cells (high Tn antigen). We observed that CARs with more HPA units exhibited higher avidity toward cells with elevated Tn antigen expression (Fig. 4f, g). However, lentiviral transduction efficiency was markedly reduced for HPAx3 CAR constructs due to the increased transgene size (Supplementary Fig. 4a). We further measured CD69 expression on CAR-T cells after a 24-h co-culture with Capan-2 and Capan-2 C1GALT1 KO cells. The dual HPA structure led to higher activation and improved in vitro killing specificity against Capan-2 C1GALT1 KO cells compared to the single HPA structure (Supplementary Fig. 4b).
We next utilized a non-immortalized pancreatic ductal adenocarcinoma patient-derived xenograft (PDX) cell line (PDX1294), which has high expression levels of both Tn antigen and mesothelin (Fig. 4h–j and Supplementary Fig. 4e). The expression level of Tn antigen on PDX1294 was higher than Capan-2 C1GALT1 KO cells or itraconazole-treated Capan-2. Confocal images confirmed that PDX1294 bind more HPA on its cell membrane than Capan-2 cells (Fig. 4h–j). The dual HPA-directed CAR-T cells exhibited enhanced cytotoxicity against PDX1294 cells than single HPA in vitro (Fig. 4j and Supplementary Fig. 4c, d). We then compared HPA-directed CAR-T cells with Tn-MUC1 CAR-T cells using PDX1294 and Capan-2 cell lines. HPA-directed CAR-T cells exhibited superior killing compared to Tn-MUC1 CAR-T cells, even in Capan-2 C1GALT1 KO cell lines, which have higher Tn-MUC1 expression (Fig. 4k–n). Using PDX1294 cells, we further evaluated triple HPA constructs to determine whether they outperform the dual HPA design. When normalized for CAR expression, the killing efficiency of the triple construct was comparable to that of the dual HPA CAR.
To explore alternative lectins that recognize Tn or sialyl-Tn (STn) antigen antigens, we next compared the performance of HPAx2 with other lectins, including the human macrophage galactose N-acetyl-galactosamine-specific lectin (MGL) and sialic acid-binding immunoglobulin-like lectin 15 (Siglec-15). MGL binds to the Tn antigen, while Siglec-15 preferentially recognizes the tumor-associated STn antigen78,79 (Supplementary Fig. 5a). Using fluorescently labeled recombinant MGL and Siglec-15, we confirmed strong binding to PDX1294 cells, indicating overexpression of both Tn and STn antigens in this model (Supplementary Fig. 5b). We then tested CAR-T cells engineered with these lectins in cytotoxicity assays. While Siglec-15 CAR-T cells exhibited minimal killing activity, MGL-based CAR-T cells showed effective cytotoxicity, with the MGLx1 construct superior killing than the dual MGL (MGLx2). However, MGLx1 CARs were still less effective than the dual HPAx2 CARs (Supplementary Fig. 5c, d). Given these comparative results, along with the large transgene size and reduced viral transduction efficiency of HPAx3, we selected the dual HPA CAR as the optimal configuration.
We then tested these dual-HPA-directed CAR-T cells in vivo using Capan2 C1GALT1 KO model. In NSG mice, HPA-directed CAR-T cells significantly reduced tumor size in Capan-2 C1GALT1 KO tumors (Supplementary Fig. 5g, h). However, in one group of mice treated with CAR T cells from Donor #1, we observed toxicity that resulted in rapid weight loss and decreased survival of these mice (Fig. 4o). We observed a similar toxicity in 1/5 mice from each of the other two donor groups, suggesting an on-target, off-tumor toxicity from the dual-HPA CAR T cells. To further investigate this toxicity, we harvested tumors (Capan-2 C1GALT1 KO) and major organs, including the kidney, lung, liver, pancreas, and spleen, from the mice and performed HPA staining. We observed notable HPA binding in the kidney and lung, suggesting potential on-target, off-tumor recognition in these tissues (Fig. 4p). This observation prompted us to repurpose the dual-HPA module as a glyco-bridge rather than using it as the primary antigen-binding domain of the CAR. Given its effective recognition of Tn antigens, HPA can serve as a modular attachment to facilitate tumor targeting without directly mediating CAR activation. These findings support the use of HPA lectins as auxiliary binders that enhance CAR function while minimizing on-target, off-tumor toxicity.
Enhancing CAR-T cell efficacy with dual HPA bridge in PDX models
We next explored the hypothesis that using a dual HPA structure as a glyco-bridge could broaden Tn antigen targeting and enhance CAR-T cell efficacy. We modified the existing glyco-bridge with the dual HPA structure (Fig. 5a–c). The expression level of CAR with HPA glyco-bridge was similar to the levels of the Tn-MUC1 bridge and CD19-bridge, as measured by (G4S)3 MFI (Supplementary Fig. 6a). To confirm that the dual HPA glyco-bridge does not induce killing, we used Jurkat cells, which overexpress Tn antigen but lack mesothelin expression (Supplementary Fig. 6b), as a control model. Even with high levels of HPA binding, glyco-bridge-mediated cytotoxicity did not occur (Supplementary Fig. 6c, d).

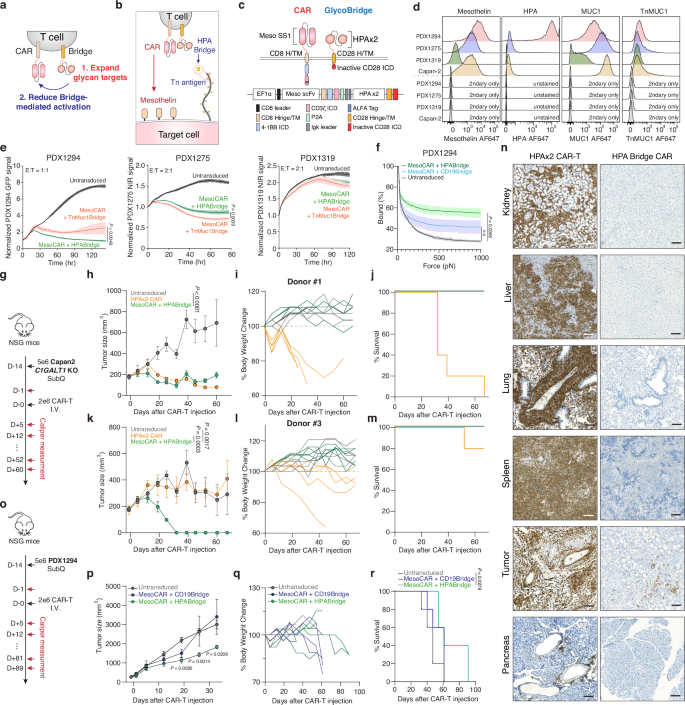
a Schematic representation of CAR-T cells with a tandem HPA bridge, showing expanded glycan targeting and reduced bridge-mediated activation. b Schematic of mesothelin-targeting CAR T cells with tandem HPAs-based glyco-bridge. c Graphic representation of CAR and bridge constructs. d Flow cytometry analysis of mesothelin, HPA, MUC1, and Tn-MUC1 expression on Capan-2 and PDX-derived cells from of n = 3 independent experimental replicates. e Real-time cytotoxicity assay against indicated PDX-derived cells with indicated CAR T cells at 1:1-2:1 E:T ratio. Results are mean ± s.d. of n = 3 independent measurements. f Strength of interaction between PDX1294 and indicated CAR-T cells. Percentage of total CAR-T cells remaining bound to target cells as the acoustic force ramp is applied from 0 to 1000 pN are shown. Results are mean ± s.e.m. of n = 3 independent measurements. g NSG mice were subcutaneously injected with 5 × 106 Capan2 C1GALT1 KO cells on day −14. On Day 0, 5 mice per group received 2 x 106 CAR-T cells with HPA bridge or HPA-directed CAR-T cell or Untransduced T cell (UTD). Tumor growth curve (h), body weight change (i) and survival percentage (j) for Capan2 C1GALT1 KO model using Donor #1. Results are mean ± s.e.m. of n = 5 mice. Tumor growth curve (k), body weight change (l) and survival percentage (m) for Capan2 C1GALT1 KO model using Donor #2. Results are mean ± s.e.m. of n = 5 mice. n Representative IHC images for human CD3 staining across different organs from Fig. 5g–m (n = 5 mice). Scale bar, 100 μm. o–r, NSG mice were subcutaneously injected with 5 × 106 PDX1294 cells on day −14. On Day 0, 5 mice per group received 2 × 106 CAR-T cells with HPA bridge or CD19-bridge or (UTD). Tumor growth curve (p), body weight change (q) and survival percentage (r) for PDX1294 cell model. Results are mean ± s.e.m. of n = 5 mice. In e, f, h, k, p, and r statistical analysis was performed by two-way ANOVA with correction for multiple comparisons.
To better evaluate this bridge-based approach, we next utilized a panel of non-immortalized patient-derived xenograft (PDX) cell lines, including the previously characterized PDX1294 and two additional models, PDX1275 and PDX1319. We first compared the expression levels of mesothelin, HPA-binding glycans, MUC1, and Tn-MUC1 across these models (Fig. 5d). PDX1294 exhibited high levels of both mesothelin and HPA-binding glycans. PDX1275 showed moderate mesothelin expression and high MUC1 expression, closely resembling the Capan-2 cell line. In contrast, PDX1319 expressed low levels of mesothelin, MUC1, and HPA-binding glycans (Fig. 5d). Using this diverse panel of PDX models, we compared the cytotoxic function of CAR-T cells equipped with either HPA-bridge or Tn-MUC1-bridge modules. As expected, PDX1294 responded more strongly to the HPA-bridge CAR-T cells, while PDX1275 was more effectively targeted by Tn-MUC1-bridge CAR-T cells. In PDX1319, limited antigen expression correlated with minimal response to both bridge strategies, including the conventional mesothelin-targeting CAR (Fig. 5e). While our shift to HPA bridge was intended to broaden the glycan recognition landscape beyond MUC1, we found that Tn-MUC1-specific constructs retained strong activity, particularly in models with high Tn-MUC1 expression but low HPA reactivity, such as PDX1275. Based on these observations, we examined whether the HPA bridge enhances CAR-T cell avidity in PDX1294, which overexpresses the Tn antigen. Similar to the Tn-MUC1 bridge shown in Fig. 1l–n, mesothelin-targeted CAR-T cells with the HPA bridge exhibited higher avidity compared to the CD19 bridge control (Fig. 5f ).
We next tested whether a dual HPA-based bridge could reduce the toxicity of HPA-CAR. Using the same donors, we conducted experiments in the Capan-2 C1GALT1 KO subcutaneous model, which was previously used for the HPA-CAR experiment in Fig. 4o, p (Fig. 5g). Interestingly, the toxicity associated with HPA-CAR was no longer present when the HPA-binding moiety was used as a bridge instead of as a CAR. Additionally, the mesothelin-targeted CAR with the HPA-bridge resulted in improved overall survival and induced either similar or greater tumor control compared to the HPA-CAR, depending on the donor (Fig. 5h–m). To explore the difference in toxicity and try to understand why the HPA-CAR was toxic, we harvested tissues from mice treated with the HPA-CAR versus the mesothelin-targeted CAR with the HPA-bridge. We found that HPA-CARs exhibited nonspecific infiltration into the kidney, liver, and lung, whereas the HPA-based bridge did not infiltrate into these organs and was primarily found in the tumor tissue (Fig. 5n and Supplementary Fig. 7). To validate this approach in another tumor model, we used a pancreatic ductal adenocarcinoma patient-derived xenograft. PDX1294 cells (5 × 106) were injected subcutaneously into NSG mice, and tumors were monitored for 14 days; 2 x 106 CAR-T cells were injected intravenously when the average tumor size reached approximately 200 mm3. After a single injection of CAR-T cells, tumor size was monitored via caliper measurement (Fig. 5o). We confirmed that CAR-T cells with a dual HPA bridge exhibited superior tumor-killing and extended survival compared to CAR-T cells with the control CD19-bridge (Fig. 5p–r and Supplementary Fig. 6e). These results demonstrate that the HPA bridge enhances CAR-T cell efficacy by improving tumor recognition and cytotoxicity in Tn antigen-rich PDACs.
Taken together, these data suggest that adding the glyco-bridge to CAR-T cells can overcome the glycocalyx barrier and increase target binding and killing. Moreover, the bridge system’s ability to activate CAR-T cells based on antigen density and binding affinity was validated. By using HPA lectin, which specifically recognizes Tn antigens regardless of their carrier glycoproteins, we incorporate a HPA lectin as a binder to the glyco-bridge module that not only improves killing efficacy, even in a pancreatic cancer PDX model, but also significantly reduces the toxicity observed from using the same binder in CAR format.
link